The impact of telomere and telomerase dysfunction on human cancer
 The upregulation of telomerase reverse transcriptase (TERT), the protein component of the enzyme that replenishes telomeres, is a key driver mutation in melanoma, glioblastoma and many other types of human malignancies. Indeed, promoter mutations in Tert are more prevalent than p53 mutations. Work from our lab and others established that telomerase function is necessary for the long-term survival of human cancer cells (Zhang et al., Genes Dev 1999; Taboski et al., Cell Rep 2012), and there is intense interest in the development of telomerase inhibitors as a treatment for cancer. At present, there are no telomerase inhibitors approved for clinical use, and there is a dearth of information about the potential emergence of drug resistance to telomerase inhibitors. We have used biochemical, genetic and chemical means to investigate the structural and functional mechanisms by which telomerase promotes survival of human cancers, and to study the possibility of resistance to telomerase inhibition.
The upregulation of telomerase reverse transcriptase (TERT), the protein component of the enzyme that replenishes telomeres, is a key driver mutation in melanoma, glioblastoma and many other types of human malignancies. Indeed, promoter mutations in Tert are more prevalent than p53 mutations. Work from our lab and others established that telomerase function is necessary for the long-term survival of human cancer cells (Zhang et al., Genes Dev 1999; Taboski et al., Cell Rep 2012), and there is intense interest in the development of telomerase inhibitors as a treatment for cancer. At present, there are no telomerase inhibitors approved for clinical use, and there is a dearth of information about the potential emergence of drug resistance to telomerase inhibitors. We have used biochemical, genetic and chemical means to investigate the structural and functional mechanisms by which telomerase promotes survival of human cancers, and to study the possibility of resistance to telomerase inhibition.
Our lab has used yeast and human cell models to biochemically characterize small molecule telomerase inhibitors (Wong et al., Chem Biol 2013). A current and complementary thrust in the lab is to understand how drug resistance to telomerase may develop in human cancer. The telomerase reverse transcriptase (RT) has structural similarity to HIV RT and thus may acquire mutations that permit drug resistance. We undertook systematic mutagenesis of TERT, and a CRISPR/Cas9 base- editing approach to introduce mutations throughout the TERT gene in a blood cancer line. In follow-up biochemical analysis of purified, in vitro-produced human telomerase we have identified possible drug-resistant alleles of TERT and are testing for their ability to resist inhibition in human cell-based models (Borges and Harrington, unpublished). We are also carrying out structural and biochemical characterization of these resistant TERT variants, to understand how they affect the catalytic properties of TERT.
Another important aspect of this research problem is the need to understand the genetic landscape of cancer cells that permit the emergence of drug resistance. Most cancer treatment strategies involving telomerase inhibition are hindered by a “therapeutic lag”, i.e. a delay in the onset of cell death until telomeres are critically eroded (Taboski et al., Cell Rep 2012). Using CRISPR/Cas9 screening technology, we characterized the genetic landscape of genes that are essential for survival in the presence of the telomerase inhibitor BIBR1532. These screens identified several new candidate genes whose deletion sensitizes cancer cells to telomerase inhibition, including TAPR1 (telomere attrition and p53 response protein 1) (Benslimane et al., Aging Cell 2021). Our data suggests that TAPR1 may act through the E3 ubiquitin ligase HUWE1 to attentuate p53 activation via increased proteolysis in response to stressors included eroded telomeres. We are further characterizing the biochemical activities of TAPR1 and other new targets identified in this screen. It is possible that inhibition of these gene targets might reduce the therapeutic lag period incurred upon telomerase inhibition (particularly in cells with longer average telomere lengths), which could have therapeutic benefit in the treatment of cancer.
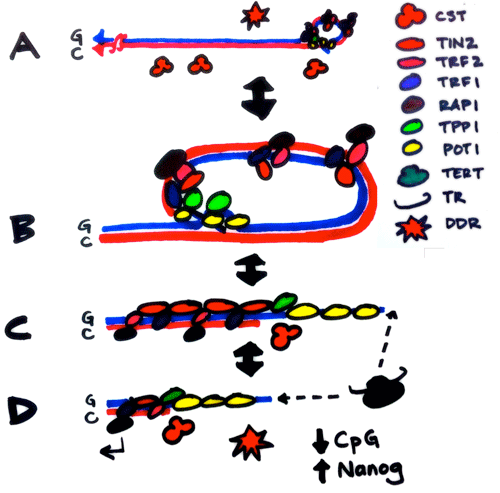
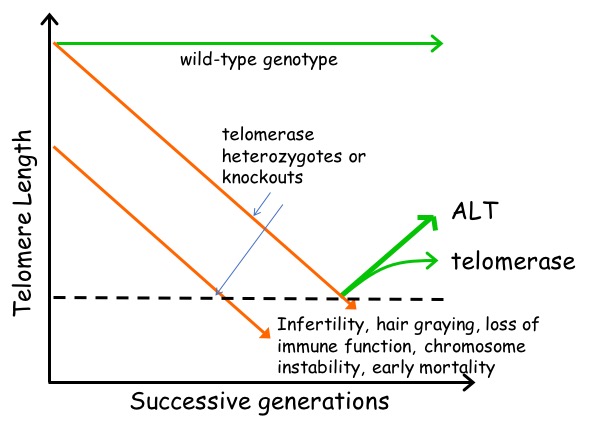
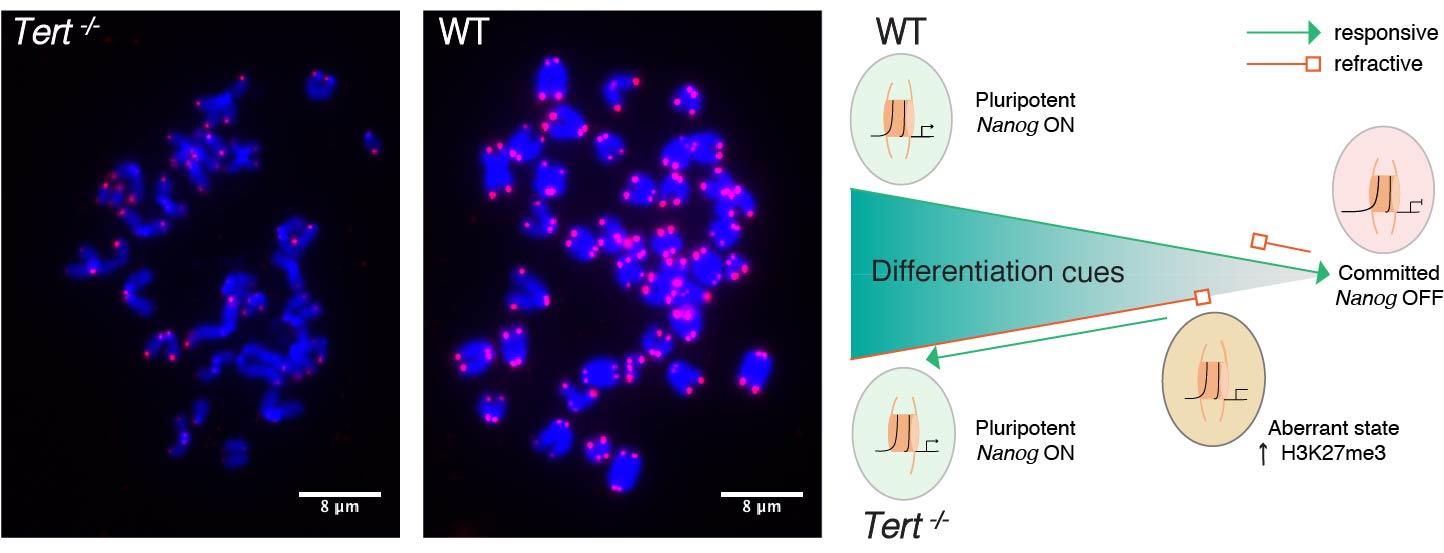 Telomerase knockout mice (e.g. see Liu et al., Curr Biol 2000; Liu et al., PNAS 2002; Erdmann et al., PNAS 2004; Meznikova et al., Dis Model Mech 2009; Erdmann et al., DNA Repair 2009) serve as a valuable in vivo model for understanding human telomeropathies that include idiopathic pulmonary fibrosis, aplastic anemia, dyskeratosis congenita, or myeloid and other cancers. Using Tert knockout embryonic stem cells, we uncovered an unexpected effect of critically short telomeres on the epigenome and the ability of stem cells to achieve and maintain a stable, differentiated state (Pucci et al., Cell Stem Cell 2013). It was known that many epigenetic alterations in DNA methylation and histone post-translational modification are hallmarks of the aging process, and that short telomeres are also a hallmark of cancer and aging, but whether there was a direct link between these phenomena was unknown. We discovered that eroded telomeres in murine ESCs impair their ability to fully consolidate a differentiated phenotype in response to all-trans retinoic acid (ATRA) (Pucci et al., Cell Stem Cell 2013). This impairment is associated with a reduction in DNA methylation and H3K27me3 levels at the Nanog promoter that leads to a failure to completely downregulate factors that promote pluripotency. More recently, we showed that chemical inhibition of the Polycomb Repressive Complex 2 (PRC2), an H3K27 tri-methyltransferase, exacerbated the differentiation impairment of mESCs when telomeres were critically eroded. Transcriptional and chromatin accessibility profiling of cells marked with a fluorescent pluripotency reporter (Oct4-GFP), uncovered a striking propensity of differentiated mESCs with short telomeres to re-acquire a pluripotent-like state. Unlike PRC2 inhibition, decreasing H3K27me3 demethylation via inhibition of Kdm6a/b led to a partial rescue of the phenotype. This data revealed a new interdependent relationship between H3K27me3 and telomere integrity in stem cell lineage commitment that may have implications in aging and cancer (Criqui et al, eLife, 2020). Currently, we are further pursuing these findings to examine the role of p53 and p21 in this differentiation defect, and whether the propensity to re-acquire pluripotent properties also occurs in aging tissues with eroded telomeres (e.g. in mice lacking Tert). Our findings suggests that an altered epigenetic state or critically eroded telomeres are not merely separate hallmarks of aged tissues and cancer, but are interconnected in important ways that affect cell function and cell fate. We wish to determine how these new mechanistic linkages between these processes can be exploited to ameliorate age-associated disease.
Telomerase knockout mice (e.g. see Liu et al., Curr Biol 2000; Liu et al., PNAS 2002; Erdmann et al., PNAS 2004; Meznikova et al., Dis Model Mech 2009; Erdmann et al., DNA Repair 2009) serve as a valuable in vivo model for understanding human telomeropathies that include idiopathic pulmonary fibrosis, aplastic anemia, dyskeratosis congenita, or myeloid and other cancers. Using Tert knockout embryonic stem cells, we uncovered an unexpected effect of critically short telomeres on the epigenome and the ability of stem cells to achieve and maintain a stable, differentiated state (Pucci et al., Cell Stem Cell 2013). It was known that many epigenetic alterations in DNA methylation and histone post-translational modification are hallmarks of the aging process, and that short telomeres are also a hallmark of cancer and aging, but whether there was a direct link between these phenomena was unknown. We discovered that eroded telomeres in murine ESCs impair their ability to fully consolidate a differentiated phenotype in response to all-trans retinoic acid (ATRA) (Pucci et al., Cell Stem Cell 2013). This impairment is associated with a reduction in DNA methylation and H3K27me3 levels at the Nanog promoter that leads to a failure to completely downregulate factors that promote pluripotency. More recently, we showed that chemical inhibition of the Polycomb Repressive Complex 2 (PRC2), an H3K27 tri-methyltransferase, exacerbated the differentiation impairment of mESCs when telomeres were critically eroded. Transcriptional and chromatin accessibility profiling of cells marked with a fluorescent pluripotency reporter (Oct4-GFP), uncovered a striking propensity of differentiated mESCs with short telomeres to re-acquire a pluripotent-like state. Unlike PRC2 inhibition, decreasing H3K27me3 demethylation via inhibition of Kdm6a/b led to a partial rescue of the phenotype. This data revealed a new interdependent relationship between H3K27me3 and telomere integrity in stem cell lineage commitment that may have implications in aging and cancer (Criqui et al, eLife, 2020). Currently, we are further pursuing these findings to examine the role of p53 and p21 in this differentiation defect, and whether the propensity to re-acquire pluripotent properties also occurs in aging tissues with eroded telomeres (e.g. in mice lacking Tert). Our findings suggests that an altered epigenetic state or critically eroded telomeres are not merely separate hallmarks of aged tissues and cancer, but are interconnected in important ways that affect cell function and cell fate. We wish to determine how these new mechanistic linkages between these processes can be exploited to ameliorate age-associated disease. 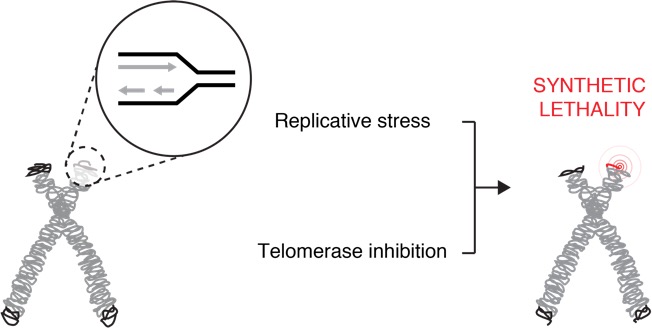 There has been a recent explosion of knowledge regarding the genetic factors that influence aging in worms, flies, yeast and mammals. Many of the gene deletions or mutations that affect lifespan in worms, for example, through perturbations in insulin signaling, also affect lifespan when mutated or altered in mice. In yeast, genome-wide screens revealed that many single gene deletions impact replicative lifespan, either positively or negatively, but the impact on aging when these gene deletions are combined is complex, which underscores that aging is a complex quantitative genetic trait. Thus, we must take advantage of all tools at our disposal—biochemical, chemical and genetic—to begin to parse out the complex properties that typify the aging process. We have taken a genome-wide approach to map the landscape of chemical-genetic interactions in response to treatment with senotherapeutics (compounds that extend lifespan in model organisms) or senolytics (compounds that lead to clearance of senescent cells), collectively referred to as seno-modulators. Using this approach, we found that the stilbenes resveratrol or pterostilbene, which extend lifespan in worms and yeast, negatively impact cell fitness of human cells via replication stress (Benslimane et al., Mol Cell, 2020). These results suggest there is much yet to learn about the genetic impact of senomodulators on cell and organismal aging. Our results may also lead us to putative new seno-modulators whose mechanism-of-action we can probe further using cell-based models and for their effects on improving the health of prematurely aging Tert knockout mice. We also plan to exploit the fact that Oct4 expression does not normally occur in adult somatic tissues, but can be re-acquired in cells with eroded telomeres after differentiation induction (Criqui et al., eLife 2020). We are currently investigating whether aged tissues (especially in a context where telomeres are eroded) exhibit an inappropriate re-expression of Oct4 or other pluripotency factors. If so, we can potentially exploit this property using specific genetic or chemical ablation strategies that will selectively remove Oct4+ positive cells from tissues, to determine if the removal of these inappropriately rewired cells can be used to improve tissue function.
There has been a recent explosion of knowledge regarding the genetic factors that influence aging in worms, flies, yeast and mammals. Many of the gene deletions or mutations that affect lifespan in worms, for example, through perturbations in insulin signaling, also affect lifespan when mutated or altered in mice. In yeast, genome-wide screens revealed that many single gene deletions impact replicative lifespan, either positively or negatively, but the impact on aging when these gene deletions are combined is complex, which underscores that aging is a complex quantitative genetic trait. Thus, we must take advantage of all tools at our disposal—biochemical, chemical and genetic—to begin to parse out the complex properties that typify the aging process. We have taken a genome-wide approach to map the landscape of chemical-genetic interactions in response to treatment with senotherapeutics (compounds that extend lifespan in model organisms) or senolytics (compounds that lead to clearance of senescent cells), collectively referred to as seno-modulators. Using this approach, we found that the stilbenes resveratrol or pterostilbene, which extend lifespan in worms and yeast, negatively impact cell fitness of human cells via replication stress (Benslimane et al., Mol Cell, 2020). These results suggest there is much yet to learn about the genetic impact of senomodulators on cell and organismal aging. Our results may also lead us to putative new seno-modulators whose mechanism-of-action we can probe further using cell-based models and for their effects on improving the health of prematurely aging Tert knockout mice. We also plan to exploit the fact that Oct4 expression does not normally occur in adult somatic tissues, but can be re-acquired in cells with eroded telomeres after differentiation induction (Criqui et al., eLife 2020). We are currently investigating whether aged tissues (especially in a context where telomeres are eroded) exhibit an inappropriate re-expression of Oct4 or other pluripotency factors. If so, we can potentially exploit this property using specific genetic or chemical ablation strategies that will selectively remove Oct4+ positive cells from tissues, to determine if the removal of these inappropriately rewired cells can be used to improve tissue function.